Új gyógyszerszemle, 2022
US Pharm. 2022;47(10):24-30.
Az FDA meghatározása szerint az új molekuláris entitások (NME-k) olyan új gyógyszerkészítmények, amelyek hatóanyagként vegyi anyagot tartalmaznak, amelyet először az Egyesült Államokban hoztak forgalomba. A 2021–2022-ben jóváhagyott NME-k alábbi leírásai ( ASZTAL 1 ) részletezi az egyes új gyógyszerek alapvető klinikai és farmakológiai profilját, valamint a legfontosabb óvintézkedéseket és figyelmeztetéseket. Tartalmazza továbbá a kiválasztott farmakokinetikai, mellékhatások, gyógyszerkölcsönhatások és adagolási adatok rövid összefoglalását, amelyeket az FDA-nak nyújtottak be a gyártó új gyógyszerkérelmének alátámasztására. Ez az áttekintés célja, hogy inkább objektív, semmint tartalmilag értékelő legyen. Az egyes NME-kre vonatkozó információkat elsősorban az FDA jóváhagyása előtt közzétett forrásokból szerezték be. A tapasztalatok egyértelműen azt mutatják, hogy az NME terápiás profiljának számos aspektusa nem derül ki a forgalomba hozatal előtti klinikai vizsgálatok során, és azután derül ki, hogy a gyógyszert szélesebb populációban alkalmazzák. Például egyes NME-k esetében a korábban nem bejelentett mellékhatások a forgalomba hozatalt követő néhány éven belül nyilvánvalóvá válnak. Egyes NME-k végül legalább egy fekete doboz figyelmeztetést kaphatnak a súlyos gyógyszermellékhatások miatt, vagy kivonják őket a forgalomból olyan biztonsági okokból, amelyeket a jóváhagyáskor nem ismertek fel. Ezért, bár ez az áttekintés néhány új gyógyszer bevezetését kínálja, alapvető fontosságú, hogy a szakemberek tisztában legyenek a terápiás profiljukban bekövetkezett változásokkal, amint azt a gyógyszerészeti szakirodalom és a betegek jelentették.
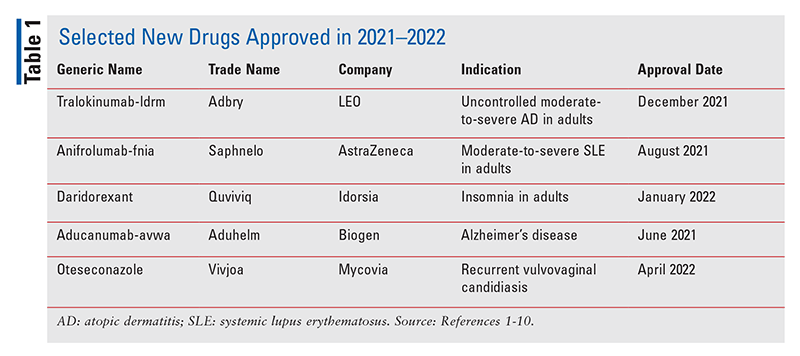
Tralokinumab-ldrm (Adbry, LEO)
Indikáció és klinikai profil 1.2 : Az FDA jóváhagyta a tralokinumab-ldrm-et közepesen súlyos vagy súlyos atópiás dermatitisz (AD) kezelésére olyan felnőtteknél, akiknek a betegsége nem kontrollálható jól helyi gyógyszerekkel, vagy ha ezek a terápiák nem tanácsosak. Kr. u., más néven atópiás ekcéma , egy krónikus, kiújuló gyulladásos bőrbetegség, amelyet száraz bőr, viszketés (különösen éjszaka) és vöröstől barnásszürkéig terjedő foltok jellemeznek a test különböző részein. Az AD incidenciája közel háromszorosára nőtt az iparosodott országokban, világszerte a gyermekek 15-20%-át és a felnőttek 1-3%-át érinti. A Tralokinumab-ldrm, az első olyan biológiai szer, amelyet az AD kezelésére hagytak jóvá, az interleukin (IL)-13 gátlásával működik, amely a betegség kulcsfontosságú gyulladásos mediátora. A Tralokinumab-ldrm helyi kortikoszteroidokkal együtt vagy anélkül is alkalmazható.
A gyógyszer jóváhagyása három kettős-vak, placebo-kontrollos, multinacionális III. fázisú vizsgálat (ECZTRA 1, ECZTRA 2 és ECZTRA 3) hatékonysági és biztonságossági eredményein alapult, amelyekben több mint 1500 felnőtt beteg vett részt. A betegek minden második héten 300 mg tralokinumab-ldrm injekciót kaptak, önmagában vagy helyi kortikoszteroidokkal együtt. Az elsődleges végpontok az Investigator's Global Assessment pontszáma 0 vagy 1 (IGA 0/1) voltak a 16. héten, és ³75%-os javulás az ekcéma területi és súlyossági indexében (EASI 75) a 16. héten. Az IGA 0/1 és/vagy Az EASI 75-öt tralokinumab-ldrm-mel a 16. héten újra randomizálták tralokinumab-ldrm-re 2-4 hetente vagy placebóra 36 hétig. A 16. héten több tralokinumab-ldrm-es beteg ért el IGA 0/1-et (38,9% vs. 26,2%) és EASI 75-öt (56,0% vs. 35,7%), mint placebót. A 16. héten reagálók közül a kéthetente tralokinumab-ldrm-mel kezeltek 89,6%-a és 92,5%-a, a 4 hetente tralokinumab-ldrm-mel kezeltek 77,6%-a, illetve 90,8%-a tartotta fenn az IGA 0/1-et és az EASI-t. 75 a 32. héten. Azoknál a betegeknél, akik nem érték el az IGA 0/1-et és az EASI 75-öt tralokinumab-ldrm-mel a 16. héten, 30,5%, illetve 55,8% érték el ezeket a végpontokat a 32. héten.
Farmakológia és farmakokinetika 1.2 : A Tralokinumab-ldrm egy humán immunglobulin (Ig) G4 monoklonális antitest, amely specifikusan kötődik a humán IL-13-hoz, és gátolja annak kölcsönhatását e receptor alfa-1 és alfa-2 alegységeivel. Az IL-13, egy tipikusan 2-es típusú immunválasz által termelt citokin, elősegíti a proinflammatorikus citokinek, kemokinek és az AD-ben szerepet játszó IgE felszabadulását.
A tralokinumab-ldrm abszolút biohasznosulása a becslések szerint 76%, a maximális plazmakoncentráció eléréséhez szükséges idő (T max ) 5-8 nappal a beadást követően. Az eloszlási térfogat becslések szerint 4,2 l. Úgy tűnik, hogy a tralokinumab-ldrm peptidmetabolizmuson megy keresztül. Felezési ideje 3 hét, a szisztémás clearance pedig 0,149 l/nap.
Mellékhatások és gyógyszerkölcsönhatások 1.2 : A klinikai vizsgálatok során a tralokinumab-ldrm-mel kapcsolatban jelentett gyakori mellékhatások közé tartozott az eozinofília, a szem és a szemhéjgyulladás, a felső légúti fertőzések és az injekció beadásának helyén fellépő reakciók. A betegeknek azt tanácsolják, hogy jelentsék egészségügyi szolgáltatójuknak az újonnan jelentkező vagy súlyosbodó szemtüneteket, mint például a kötőhártya-gyulladás vagy keratitis. Súlyos túlérzékenységi reakciók – beleértve az anafilaxiát – és angioödéma fordultak elő, amelyek miatt a gyógyszer abbahagyása volt szükséges. A tralokinumab-ldrm veszélyeztetheti az antihelminth terápiák hatékonyságát; ezért a kezelést nem szabad elkezdeni vagy folytatni olyan betegeknél, akik már fennálló helminth fertőzésben szenvednek, vagy akik a kezelés során fertőződnek meg. A tralokinumab-ldrm terápia során kerülni kell az élő vakcinák alkalmazását.
A gyógyszerkölcsönhatások tekintetében egy kis klinikai vizsgálatban a tralokinumab-ldrm hatását értékelték a midazolám (CYP3A4 szubsztrát), warfarin (CYP2C9 szubsztrát), omeprazol (CYP2C19 szubsztrát), metoprolol (CYP2D6 szubsztrát) és koffein YP1A2 szubsztrát farmakokinetikájára. nincs jelentős változás az expozícióban (AUC vagy C max ) a CYP450 szubsztrátjainak aránya többszörös tralokinumab beadás után, összehasonlítva az önmagában adott tralokinumab-ldrm-mel.
Adagolás és adminisztráció 1.2 : A Tralokinumab-ldrm 150 mg/ml-es előretöltött fecskendőben kapható SC injekcióhoz. Az ajánlott adag 600 mg kezdő adag (négy 150 mg-os injekció), majd 300 mg (két 150 mg-os injekció) kéthetente beadva. 16 hetes kezelés után a 100 kg-nál kisebb testtömegű betegeknél 4 hetente 300 mg-os adag megfontolható, ha bőre tiszta vagy majdnem tiszta. A jelenlegi immunizálási irányelvek szerint az életkornak megfelelő védőoltásokat a kezelés megkezdése előtt el kell végezni.
Anifrolumab-fnia (Saphnelo, AstraZeneca)
Indikáció és klinikai profil 3.4 : Az anifrolumab-fnia egy I-es típusú interferon (IFN) receptor antagonista, amelyet közepesen súlyos vagy súlyos szisztémás lupus erythematosus (SLE) kezelésére hagytak jóvá standard terápiában részesülő felnőtteknél. Az anifrolumab-fnia biztonságosságát és hatásosságát három multinacionális, placebo-kontrollos klinikai vizsgálatban állapították meg: MUSE, TULIP-1 és TULIP-2. A MUSE egy II. fázisú vizsgálat volt, amelynek elsődleges végpontjai az SLE válaszadó index (SRI-4) felmérése és a 24. héten mért orális kortikoszteroidok (OCS) tartós csökkenése volt. Összesen 305 beteget randomizáltak 1:1:1 arányban anifrolumab kezelésre. -fnia 300 mg, anifrolumab-fnia 1000 mg, vagy placebo. A III. fázisú TULIP-1 és TULIP-2 vizsgálatok elsődleges eredménye a betegségaktivitás javulása volt az 52. héten, amelyet SRI-4-gyel mértek a TULIP-1-ben és a British Isles Lupus Assessment Group alapú kompozit lupusvizsgálattal (BICLA) TULIP-2. A TULIP-1 és a TULIP-2 másodlagos eredménye az OCS-csökkenés fenntartása, a bőr SLE aktivitásának javulása és a fellángolási arány.
A BICLA válaszarányát az 52. héten mindhárom vizsgálatban jelentették, de ez csak a TULIP-2 esetében volt az elsődleges végpont. A válaszadási arányok közötti különbség a MUSE esetében 28,8%, a TULIP-1 esetében 17%, a TULIP-2 esetében 16,3% volt. Az SRI-4 válaszarány az 52. héten 24% volt a MUSE, 6% a TULIP-1 és 18,2% a TULIP-2 esetében. Az anifrolumab-fnia nem mutatott statisztikailag szignifikáns különbséget a TULIP-1 válaszarányában.
Farmakológia és farmakokinetika 3.4 : Az anifrolumab-fnia egy immunglobulin (Ig) G1-kappa monoklonális antitest, amely az I. típusú IFN receptor (IFNAR1) 1. alegységéhez kötődik, gátolva annak aktivitását. Az I-es típusú IFN-ek aktivitásának csökkenése csökkenti szerepüket az SLE patogenezisében. Az anifrolumab-fnia az IFNAR1 internalizálására is hat, csökkentve a rendelkezésre álló receptorokat. Ezek a hatásmechanizmusok csökkentik a gyulladást és az immunológiai folyamatokat, amelyek hozzájárulnak az SLE patológiájához.
Az anifrolumab-fnia nemlineáris farmakokinetikát mutat a 100 mg és 1000 mg közötti 4 hetente adagolt dózistartományban, ami a dózis növelésével nagyobb AUC-expozíciót eredményez. A tipikus SLE-betegben (69,1 kg) a megoszlási térfogat becslések szerint 6,23 l. A standard adagolás alapján a 4 hetente adott 300 mg anifrolumab-fnia clearance-e 0,193 l/nap.
Mellékhatások és gyógyszerkölcsönhatások 3.4 : Az anifrolumab-fnia leggyakoribb (>10%) mellékhatásai a fertőzések voltak (a betegek 70%-a), beleértve az influenzát, a húgyúti fertőzést, a tuberkulózist, a hörghurutot és (az incidencia 34%-a) a felső légúti fertőzéseket (azaz nasopharyngitis, pharyngitis). és arcüreggyulladás). A súlyos fertőzések előfordulása a kezelés során 5% volt. Jelenleg az anifrolumab-fnia emberi terhességben történő alkalmazására vonatkozó adatok korlátozottak, és nem elegendőek ahhoz, hogy tájékoztassák a súlyos születési rendellenességek, a vetélés vagy a káros anyai vagy magzati kimenetel gyógyszerrel összefüggő kockázatát. Ismeretes, hogy a terhesség előrehaladtával a monoklonális IgG antitestek aktívan átjutnak a placentán; ezért a magzat anifrolumab-fnia expozíciója nagyobb lehet a harmadik trimeszterben.
Az anifrolumab-fnia egyetlen jelentett ellenjavallata az anifrolumabbal vagy a készítmény bármely összetevőjével szembeni súlyos túlérzékenység (pl. anafilaxia). Klinikailag jelentős, aggodalomra okot adó mellékhatások a súlyos fertőzések, túlérzékenységi reakciók, beleértve az anafilaxiát és a rosszindulatú daganatok. A mai napig nem végeztek hivatalos gyógyszer-kölcsönhatási vizsgálatokat.
Adagolás és adminisztráció 3.4 : Az Anifrolumab-fnia 300 mg/2 ml (150 mg/ml) egyadagos injekciós üvegben kerül forgalomba. Az ajánlott adag 300 mg iv. infúzióban, 30 perc alatt. Beadás előtt az anifrolumab-fniát 0,9%-os nátrium-klorid injekcióval (USP) kell hígítani, aszeptikus technikával. 2 ml térfogatú 0,9%-os nátrium-klorid injekciót veszünk ki egy 100 ml-es infúziós zsákból, és enyhe keverés közben 2 ml anifrolumab-fniát adunk hozzá úgy, hogy a teljes térfogat 100 ml legyen. Az infúziós oldatot közvetlenül az elkészítést követően, 30 percen keresztül kell beadni egy steril, alacsony fehérjekötődésű 0,2 vagy 0,22 mikronos beépített szűrőt tartalmazó infúziós vezetéken keresztül. Az infúzió befejezése után a vezetéket át kell öblíteni 25 ml 0,9%-os nátrium-klorid injekcióval, USP.
Daridorexant (Quviviq, Idorsia)
Indikáció és klinikai profil 5.6 : A daridorexant olyan felnőttek kezelésére engedélyezték, akiknél az alvászavar és/vagy az alvás fenntartása nehézségekkel jellemezhető. Az álmatlanság, a leggyakoribb alvászavar (amely az Egyesült Államokban élő felnőttek több mint 30%-ánál fordul elő), az elegendő alvás nehézségét és az alvással való elégedetlenséget jelenti, amely jelentős negatív hatással van a nappali funkcióra. A krónikus álmatlanság az alvás megkezdésének és/vagy alvásának legalább 3 hónapig tartó, legalább 3 éjszakán át történő fenntartásának nehézsége. Az álmatlanság, amely a jelek szerint az ébrenléttel összefüggő agyterületek túlműködéséből adódik, jelentős hatással van a szenvedő hangulatára, energiaszintjére és koncentrációs képességére. Ezenkívül a hosszú távú álmatlanság súlyos egészségügyi állapotokkal, például pszichiátriai rendellenességekkel, szív- és érrendszeri betegségekkel, 2-es típusú cukorbetegséggel, kábítószer-használattal és demencia társul.
A daridorexant hatékonyságát két egyformán randomizált, többközpontú, placebo-kontrollos vizsgálatban értékelték: 1. vizsgálat és 2. vizsgálat. Összesen 1854 álmatlanságban szenvedő alany vett részt a Mentális Betegségek Diagnosztikai és Statisztikai kézikönyve , 5. kiadás, 3 hónapon keresztül naponta egyszer, este kapott daridorexant vagy placebót. Az 1. vizsgálatban 930 alany 25 mg daridorexant, 50 mg daridorexant vagy placebót kapott; A 2. vizsgálatban 924 alany 25 mg daridorexant, 10 mg daridorexant vagy placebót kapott. A 3 hónapos kezelési időszak végén mindkét vizsgálat tartalmazott egy 7 napos placebo-kifutó periódust, amely után az alanyok egy kiterjesztett vizsgálatba léphettek. Összesen 600 alanyt kezeltek legalább 6 hónapig; közülük 373-at legalább 12 hónapig kezeltek. Mindkét vizsgálat elsődleges hatékonysági végpontja a kiindulási állapottól az 1. és 3. hónapig a tartós alvásig (LPS, elalvásig indukcióig eltelt idő) és az elalvás utáni ébrenlét (WASO, alvásfenntartás) latenciájának változása volt, objektíven, egy alváslaboratóriumban végzett poliszomnográfiával mérve. Az 1. vizsgálatban a 25 mg-os daridorexant és az 50 mg-os daridorexant statisztikailag szignifikáns javulást mutatott a placebóhoz képest az LPS és a WASO, valamint az önbeszámolt teljes alvási idő (sTST) tekintetében az 1. és a 3. hónapban. Az 50 mg-os dózis a napközbeni idő szignifikáns csökkenését is kimutatta. álmosság a placebóval szemben – az álmossági tartomány pontszámával mérve az Insomnia Daytime Symptoms and Impacts Questionnaire 7. kérdőívéből – az 1. és 3. hónapban, amely kulcsfontosságú másodlagos végpont. A 2. vizsgálatban a 25 mg-os dózis statisztikailag szignifikáns javulást mutatott a placebóhoz képest a WASO-ban és az sTST-ben az 1. és 3. hónapban, de a 10 mg-os dózis nem.
Farmakológia és farmakokinetika 5.6 : Az orexin A és az orexin B neuropeptidek, amelyeket a neuronok választanak ki, főleg az oldalsó hipotalamuszban. Ezek a neuropeptidek két G-protein-kapcsolt receptorhoz (GPCR) kötődve és aktiválva váltják ki hatásukat: az 1-es típusú orexin receptorhoz és a 2-es típusú orexin receptorhoz (OX1R, OX2R). Az orexin receptor útvonalak létfontosságú szabályozó szerepet játszanak számos élettani folyamatban, beleértve az alvás-ébrenlét ritmust is. Daridorexant ( 1.ÁBRA ) egy benzimidazol-pirrolidin, amelyet úgy terveztek, hogy az OX1R és az OX2R kettős antagonistájaként működjön, ezáltal elnyomja az orexin A és orexin B ébredést elősegítő aktivitását.

A daridorexant orális biohasznosulása 62%, a plazma csúcskoncentrációja 1-2 órán belül alakul ki (T max ). A daridorexant eloszlási térfogata 31 liter, és 99,7%-ban kötődik a plazmafehérjékhez. A daridorexant kiterjedt metabolizmuson megy keresztül, elsősorban a CYP3A4 (89%) által. A kiválasztás elsődleges módja a széklettel (57%); kisebb rész (28%) választódik ki a vizelettel, elsősorban metabolitok formájában. A daridorexant terminális felezési ideje körülbelül 8 óra. A daridorexant gyors felszívódása az elalváshoz, valamint a kiürülési profilja, amely egy éjszakai alvás után 80%-os eliminációt eredményez, minimálisra csökkentheti a fennmaradó hatásokat.
Mellékhatások és gyógyszerkölcsönhatások 5.6 : A klinikai vizsgálatok során a daridorexanttal kapcsolatos leggyakoribb mellékhatások (>5%, és nagyobb, mint a placebónál) a fejfájás és az aluszékonyság vagy a fáradtság voltak. A termék címkéje figyelmeztetést is tartalmaz a súlyosbodó depresszióra és az öngyilkossági gondolatokra, valamint az olyan összetett alvási viselkedésekre, mint az alvajárás, az alvás közbeni vezetés és a nem teljesen ébren végzett tevékenységek (pl. ételkészítés/evés, telefonálás, szex). . A betegeknek haladéktalanul kapcsolatba kell lépniük egészségügyi szolgáltatójukkal, ha ezen káros hatások bármelyikét észlelik. Több percig tartó hallucinációk és alvási bénulás (átmeneti mozgás- vagy beszédképtelenség) fordulhatnak elő, amikor a beteg elalszik vagy felébred. A Daridorexant ellenjavallt narkolepsziában szenvedő betegeknél.
Nem állnak rendelkezésre adatok a daridorexant terhes nőkön történő alkalmazásáról a súlyos születési rendellenességek, vetélés vagy egyéb káros anyai vagy magzati kimenetelek gyógyszerrel összefüggő kockázatának meghatározására. Állatokon végzett reprodukciós vizsgálatokban a daridorexant nem okozott magzati toxicitást a maximálisan ajánlott humán dózis nyolc-tízszeresét meghaladó adagokban. Nincs adat a daridorexant anyatejben való jelenlétéről, a szoptatott csecsemőre gyakorolt hatásáról vagy a tejtermelésre gyakorolt hatásáról, de a gyógyszert és metabolitjait kimutatták szoptató rágcsálók tejében. Ezért az anyatejjel potenciálisan daridorexantnak kitett csecsemőket ellenőrizni kell a túlzott szedáció szempontjából. A daridorexant szövetségileg szabályozott anyag, mivel visszaélhető, és függőséget okozhat.
Adagolás és adminisztráció 5.6 : A Daridorexant 25 mg-os és 50 mg-os filmtablettaként kerül forgalomba orális adagolásra. Az ajánlott adag 25-50 mg naponta egyszer, lefekvés előtt 30 percen belül, és legalább 7 órával a tervezett ébredés előtt. Az elalvás kezdete késhet, ha a daridorexant étkezés közben vagy röviddel étkezés után veszi be. A Daridorexant nem szedhető más központi idegrendszeri depresszánsokkal vagy alkohollal együtt. A betegeket azt is tanácsolni kell, hogy kerüljék a gépjárművezetést, nehézgépek kezelését vagy bármilyen olyan tevékenység végzését, amely tiszta gondolkodást igényel, miközben a gyógyszer hatásait még tapasztalják. Közepesen súlyos májkárosodásban szenvedő betegeknél a maximális ajánlott adag 25 mg, legfeljebb egyszer éjszakánként. A Daridorexant nem javasolt súlyos májkárosodásban szenvedő betegeknek. Kerülni kell a daridorexant és a CYP3A4 erős inhibitoraival való egyidejű alkalmazását, és az adagot 25 mg-ra kell korlátozni azoknál a betegeknél, akik egyidejűleg mérsékelt CYP3A4-inhibitorokkal kezelnek. A daridorexant mérsékelten vagy erős CYP3A4 induktorokkal történő egyidejű alkalmazása szintén kerülendő.
Aducanumab-avwa (Aduhelm, Biogen)
Indikáció és klinikai profil 7.8 : Az Aducanumab-avwa egy amiloid-béta (AB) által irányított antitest, amelyet Alzheimer-kór kezelésére javallottak, és ez az első új kezelés e betegség kezelésére 2003 óta. terápiás előnyt jelent a súlyos vagy életveszélyes betegségek meglévő kezeléseivel szemben. Ennek a javallatnak a további jóváhagyása a klinikai előnyök megerősítő vizsgálatok során történő igazolásától függ. Az Alzheimer-kór, egy visszafordíthatatlan, legyengítő és progresszív agyi rendellenesség, amely 6,2 millió amerikait érint, lassan erodálja a memóriát, a gondolkodást és – végül – az egyszerű feladatok elvégzésének képességét is. Bár nem teljesen ismert, hogy pontosan mi okozza, az Alzheimer-kórt agyi elváltozások jellemzik, beleértve az amiloid plakkokat és a neurofibrilláris (tau) gubancokat, amelyek a memóriát és a gondolkodást szabályozó kritikus agyi régiókban neuronok elvesztését eredményezik. A jelenleg elérhető terápiák csak a betegség tüneteit kezelik, de az aducanumab-avwa az első olyan szer, amely megcélozza és megváltoztatja az Alzheimer-kór hátterében álló feltételezett folyamatot.
Az aducanumab-avwa gyorsított jóváhagyása három kettős-vak, placebo-kontrollos, dózistartományos vizsgálat eredményein alapult, amelyekben összesen 3482 enyhe kognitív károsodásban vagy Alzheimer-kór enyhe demencia stádiumában szenvedő beteg vett részt. Az EMERGE (1. vizsgálat), ENGAGE (2. vizsgálat) és PRIME (3. vizsgálat) néven ismert tanulmányok pozitronemissziós tomográfiát alkalmaztak az Alzheimer-kór patológiájában szerepet játszó agyi régiókban az AB plakkok számszerűsítésére, szemben a betegségben nem érintett agyi régiókkal. Az aducanumab-avwa-t kapó betegeknél szignifikáns dózisfüggő és időfüggő AB plakk csökkenés volt megfigyelhető (a vizsgálatok során 59%-71%), míg a kontrolloknál nem csökkent az AB plakk mennyisége. A mai napig nem állnak rendelkezésre biztonságossági vagy hatásossági adatok az aducanumab-avwa-kezelés megkezdésére vonatkozóan az Alzheimer-kór korábbi vagy későbbi szakaszában.
Farmakológia és farmakokinetika 7.8 : Az Aducanumab-avwa egy humán immunglobulin-gamma (IgG) 1 monoklonális antitest, amely képes átjutni a vér-agy gáton, hogy szelektíven megcélozza és megkösse az aggregált oldható oligomereket és az agyban lévő AB plakkok oldhatatlan fibrillumait. Biokémiai vizsgálatok kimutatták, hogy az aducanumab-avwa szelektíven kötődik a 3-7. AB aminosavak által alkotott lineáris epitóphoz, ezáltal minimálisra csökkenti a más fehérjékkel való kölcsönhatásokat. Gyenge monovalens affinitása, gyors kötődési kinetikája és az epitópban gazdag aggregátumok iránti erős aviditása alapján kimutatták, hogy az aducanumab-avwa különbséget tesz az AB monomerek és az oligomer vagy fibrilláris aggregátumok között.
Az aducanumab-avwa egyensúlyi koncentrációját 16 hetes ismételt adagolással érik el, 4 hetente történő adagolással. A csúcskoncentráció (C max ), minimális koncentráció (C min ), valamint a plazmakoncentráció-idő görbe alatti terület egyensúlyi állapotban dózisarányos módon a standard adagolási tartományban (1-10 mg/kg 4 hetente). A becsült eloszlási térfogat egyensúlyi állapotban 9,6 l. Az endogén IgG-hez hasonlóan az aducanumab-avwa is feltehetően kis peptidekké hidrolizálódik, és végül aminosavakká hidrolizálódik a fehérje katabolikus utakon. Az aducanumab-avwa terminális felezési ideje körülbelül 25 nap. Vese- vagy májkárosodásban szenvedő betegek farmakokinetikáját értékelő vizsgálatokról nem számoltak be, de a gyógyszer várhatóan nem vesén keresztül ürül vagy metabolizálódik a májban. A testtömegben, életkorban, nemben és rasszban bekövetkezett eltérések a gyógyszerexpozíció megváltozását eredményezhetik, de nem találták jelentős hatással a klinikai válaszre.
Mellékhatások és gyógyszerkölcsönhatások 7.8 : Az aducanumab-avwa-val kapcsolatban leggyakrabban jelentett mellékhatások közé tartozik az amiloiddal kapcsolatos képalkotó rendellenességek (ARIA); fejfájás; esik; hasmenés; és zavartság, tájékozódási zavar és megváltozott mentális állapot. A termék címkéje figyelmeztetést tartalmaz az ARIA-ra, amely leggyakrabban az agy egyes részeinek átmeneti duzzanataként jelentkezik; ez a hatás általában idővel megszűnik, és nem ismert, hogy tüneteket okoz. Néhány ARIA-ban szenvedő beteg azonban fejfájásról, zavartságról, szédülésről, látásváltozásról vagy émelygésről számolt be, amely az aducanumab-avwának tulajdonítható. Ritkán az aducanumab-avwa alkalmazása túlérzékenységi reakciókkal járt, beleértve az angioödémát és a csalánkiütést.
Adagolás és adminisztráció 7.8 : Az Aducanumab-avwa tartósítószer-mentes, steril oldatként kerül forgalomba intravénás injekcióhoz 170 mg/1,7 ml-es (100 mg/ml) és 300 mg/3 ml-es (100 mg/ml) egyadagos injekciós üvegekben. A kezdeti titrálást követően (6 héten keresztül) az ajánlott adag 10 mg/ttkg IV infúzióban, körülbelül 1 órán keresztül, 4 hetente, legalább 21 napos időközönként. A kezdeti titrálás 1 mg/ttkg adag beadását jelenti az 1. és 2. infúzióhoz; 3 mg/ttkg adag a 3. és 4. infúzióhoz; és 6 mg/ttkg dózis az 5. és 6. infúzió esetében. Vese- vagy májkárosodás esetén nem várható dózismódosítás.
Oteseconazole (Vivjoa, Mycovia)
Indikáció és klinikai profil 9.10 : Az otesekonazol az első és egyetlen gyógyszer, amelyet kifejezetten a kiújuló vulvovaginális candidiasis (RVVC) előfordulásának csökkentésére javasoltak olyan nőknél, akiknek anamnézisében RVVC szerepel, és akik nem rendelkeznek reproduktív potenciállal. Használata ellenjavallt terhes és szoptató nőknél. Az RVVC, közismert nevén krónikus élesztőfertőzés, úgy definiálható, mint három vagy több tüneti élesztőfertőzés akut epizódja 12 hónapon belül. A tünetek közé tartozik a hüvelyi viszketés, égő érzés, irritáció és gyulladás, és néhány beteg rendellenes hüvelyváladékot és fájdalmas vizelést vagy szexuális közösülést tapasztal. Becslések szerint a felnőtt nők 75%-ánál lesz élete során legalább egy gombás fertőzés, és körülbelül 50%-uk fog kiújulni; ezeknek a nőknek akár 90%-ánál alakul ki RVVC. Az FDA megadta az oteseconazol Fast-Track és minősített fertőző betegségek termékének megjelölését.
Az otesekonazolt három, 11 ország 232 helyszínéről származó 875 beteg bevonásával végzett III. fázisú klinikai vizsgálat hatékonysági adatai alapján hagyták jóvá. A két globális VIOLET vizsgálatban az otesekonazolt kapó RVVC-ben szenvedő nők 93-96%-ánál nem fordult elő kiújulás a 48 hetes fenntartó időszakban; ezzel szemben a placebót kapók 57-61%-a nem tapasztalt kiújulást. Az egyesült államokbeli UltraVIOLET vizsgálatban az oteseconazolt kapó RVVC-ben szenvedő nők 89,7%-ánál megszűnt a kezdeti élesztőfertőzés, és nem volt kiújulása az 50 hetes fenntartó időszak alatt, míg a flukonazolt és placebót kapó nők 57,1%-ánál nem volt kiújulás.
Farmakológia és farmakokinetika 9.10 : Oteszekonazol ( 2. ÁBRA ) egy új tetrazol gombaellenes származék, amely gátolja a 14-alfa-demetilázt (CYP51), egy gomba enzimet, amely katalizálja az esszenciális gomba szterol ergoszterol bioszintézisének korai lépését. Ezenkívül a 14-alfa-demetiláz gátlása 14-metilezett szterolok felhalmozódását eredményezi, amelyek mérgezőek a gombákra. A tetrazol fémkötő csoport szerkezetébe való beépülése miatt az oteszekonazol csökkent affinitással rendelkezik a humán CYP enzimekhez, és feltehetően jobb a mellékhatás- és gyógyszerkölcsönhatás-profilja.

Az otesekonazol plazma csúcskoncentrációjának eléréséhez szükséges idő 5-10 óra. Magas zsírtartalmú, kalóriadús étellel történő beadás növelte a C-t max és AUC 0-72 óra 45%-kal, illetve 36%-kal, de nem figyeltek meg szignifikáns különbséget az alacsony zsírtartalmú, alacsony kalóriatartalmú ételeknél. Az otesekonazol eloszlási térfogata körülbelül 423 liter, és a gyógyszer >99%-ban kötődik a plazmafehérjékhez. Az állatkísérletek azt mutatják, hogy az oteszekonazolnak a hüvelyszövetben való expozíciója összevethető a plazmaexpozícióval. Az otesekonazol nem metabolizálódik jelentős mértékben. Az orális adag körülbelül 56%-a a széklettel ürül epével, 26%-a pedig a vizelettel. Az otesekonazol átlagos terminális felezési ideje körülbelül 138 nap.
Mellékhatások és gyógyszerkölcsönhatások 9.10 : A leggyakrabban jelentett mellékhatás a fejfájás és a hányinger volt, amelyek a vizsgálatban résztvevők 7,4%-ánál, illetve 3,6%-ánál fordultak elő. A klinikai vizsgálatok során a betegek <2%-ánál jelentett mellékhatások közé tartozik a dyspepsia, hőhullám, dysuria, a reproduktív rendszer rendellenességei (menorrhagia, metrorrhagia, vulvovaginális irritáció) és a vér kreatin-foszfokinázszintjének emelkedése. Állatkísérletek során az otesekonazol magzati károsodást okoz; ezért ellenjavallt szaporodási képességű nőstényeknél, valamint terhes és szoptató nőknél a magzatot vagy a szoptatott csecsemőt érintő lehetséges kockázatok miatt. Az otesekonazol ellenjavallt olyan betegeknél is, akiknél ismert túlérzékenység a gyógyszerre.
Az otesekonazol az emlőrák-rezisztencia fehérje (BCRP) inhibitora, és a BCRP szubsztrátokkal, például rozuvasztatinnal történő egyidejű alkalmazása növelheti a szubsztrát expozícióját, növelve az ezekkel a szerekkel kapcsolatos mellékhatások kockázatát. Ezért ajánlatos a BCRP szubsztrát legalacsonyabb hatásos kezdeti és fenntartó dózisát használni, ha azt oteszekonazollal egyidejűleg adják, és a beteget ellenőrizni kell a mellékhatások tekintetében.
Adagolás és adminisztráció 9.10 : Az otesekonazol 150 mg-os keményzselatin kapszula formájában kapható orális adagolásra. Két ajánlott adagolási rend létezik: 1) önmagában oteseconazol és 2) oteseconazol plusz flukonazol. A termék címkéje részletes adagolási ütemtervet tartalmaz ezekhez a sémákhoz. Enyhe-közepes vesekárosodásban vagy enyhe májkárosodásban szenvedő betegeknél az adagolás módosítása nem javasolt; az oteszekonazol azonban nem javasolt súlyos vesekárosodásban vagy végstádiumú vesebetegségben (dialízissel vagy anélkül), illetve közepesen súlyos vagy súlyos májkárosodásban szenvedő betegeknél.
IRODALOM
1. Adbry (tralokinumab-ldrm) betegtájékoztató. Madison, NJ: LEO Pharma Inc; 2022. július.
2. Wollenberg A, Howell MD, Guttman-Yassky E, et al. Az atópiás dermatitisz kezelése tralokinumabbal, egy anti-IL-13 mAb-vel. J Allergy Clin Immunol . 2019;143(1):135-141.
3. Saphnelo (anifrolumab-fnia) betegtájékoztató. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2022. július.
4. Anifrolumab: gyógyszerinformáció. Naprakész. Waltham, MA: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
5. Quviviq (daridorexant) betegtájékoztató. Radnor, PA: Idorsia Pharmaceuticals US Inc.; 2022. április.
6. Markham A. Daridorexant: első jóváhagyás. Kábítószer . 2022;82(5):601-607.
7. Aduhelm (aducanumab-avwa) betegtájékoztató. Cambridge, MA: Biogen Inc.; 2021. június.
8. Sevigny J, Chiao P, Williams L és munkatársai. Aducanumab (BIIB037), egy anti-amiloid béta monoklonális antitest prodromális vagy enyhe Alzheimer-kórban szenvedő betegeknél: egy randomizált, kettős-vak, placebo-kontrollos, 1b fázisú vizsgálat időközi eredményei. Alz demencia . 2015;11 (7. kiegészítés 6):P277.
9. Vivjoa (oteseconazole) betegtájékoztató. Durham, NC: Mycovia Pharmaceuticals, Inc.; 2022. április.
10. Sobel JD, Nyirjesy P. Oteseconazole: előrelépés a visszatérő vulvovaginális candidiasis kezelésében. Future Microbiol . 2021;16(18):1453-1461.
A cikkben található tartalom csak tájékoztató jellegű. A tartalom nem helyettesíti a szakmai tanácsokat. A cikkben közölt információkra való hagyatkozás kizárólag a saját felelősségére történik.











